A journal, first published in 1985, that focuses on developments in genome bioinformatics and computational biology. It now appears monthly.
http://bioinformatics.oxfordjournals.org/ Journal home page.
Since the 1980s molecular biology has undergone a revolution, with the introduction of automated high-throughput techniques for determining the chemical composition of biomolecules, such as DNA and proteins. Genome projects, notably the Human Genome Project, have generated vast amounts of sequence data, detailing the order of bases throughout entire genomes.
Much of the information is held in various databases hosted by public institutions in different countries. Storage, handling, and analysis of these data have been made possible by advances in computer science and information technology, creating the field of bioinformatics.
Bioinformatics developed on the founding principles of unlimited access without charge or constraint. Fortuitously, the bioinformatics revolution coincided with the rise of the Internet and the free flow of information around the world. A consequence has been the integration of data from many different sources, with an emphasis on accessibility. The resulting global bioinformatics community has proved enormously beneficial to many areas of research, including medicine, pharmacology, and forensic science, as well as biology.
Making sense of sequences
Stretches of DNA, or entire genomes of organisms, can be sequenced in a matter of hours or days. But what does the resulting string of As, Cs, Gs, and Ts mean?
The usual starting point is to compare a sequence—the query sequence—with existing sequences held on public databases. Such comparisons typically use computing methods (algorithms) and software packages developed and provided as open source (free) by the database-hosting organization.
Finding a similar sequence might provide important clues about possible function. For example, a homologous gene in another organism might have the same function as the query gene. Genes (and proteins) with important functions tend to be highly conserved even in distant relatives.
Computerized analysis of a sequence will also identify such regions of the sequence as open reading frames, promoters, enhancers, and repetitive DNA and translate any putative coding sequences into corresponding amino acid sequences. Information about the epigenetic chemical modifications to DNA and histone proteins that influence gene activity will have a bearing on predicting how a particular gene will function, e.g. in cancer cells or other disease states.
Amino acid sequences can be analysed similarly, to pinpoint possible structural features, such as DNA- or protein-binding domains, and to produce the most likely three-dimensional structures of the assembled proteins.
Comparative genomics
Thousands of organisms from all domains of the living world are now represented in sequence databases. This torrent of information is expanding our knowledge of how genomes are organized and how they work, as well as clarifying the evolutionary relationships between different groups of organisms.
Computerized genome maps are available for many organisms. These make it much easier to compare the order of genes and markers along chromosomes in different species, to establish how the genetic material evolves.
The wonderful world of ‘-omics’
Genome sequence data are just the start; they can be regarded as the ‘instruction manual’ available to a particular organism in the form of its genes, and their ability to encode proteins. Bioinformatics also encompasses data accumulating from automated analyses of all aspects of cell metabolism directed by those genes, including:
• transcriptomics—which genes are transcribed into messenger RNA molecules by different types of cells?
• proteomics—what proteins do the cells actually make, when do they make them, and in what quantities?
• interactomics—what combinations of proteins are required for particular tasks?
• epigenomics—how do epigenetic modifications to chromatin affect gene activity?
These data come from such techniques as microarrays, RNA-seq, and mass spectrometry. Integrating the data from these sources, although a hugely challenging task, is providing new insights into various aspects of how cells work during embryological development, in health and disease, why they sometimes become cancer cells, and how they respond to drugs.
A long-term goal of bioinformatics is to pave the way for accurate computerized models of entire cells or even organisms (see systems biology).
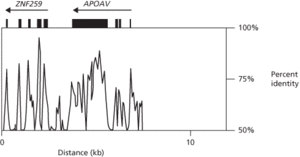
This graph shows percent identity between human and mouse DNA in the region of the APOAV gene, which encodes apolipoprotein A–V (a regulator of triglyceride levels in blood), and the ZNF259 gene, which encodes zinc finger 259. Black bars indicate exons of genes; arrows indicate direction of transcription. Note increased similarity in exons compared with introns.

Part of an alignment of amino acid sequences of an isoform of apolipoprotein A–V from human and mouse, using BLAST (Basic Local Alignment Search Tool). It enables comparison of the human sequence with the well-characterized mouse sequence and is a key step in identifying its nature, possible function, and evolutionary relationships. Dashes in human and mouse lines indicate gaps; the line between them indicates shared identical amino acids and conserved residues (i.e. chemically similar amino acids, denoted by +).
- Armstrong, David Malet (1926–2014)
- Armstrong Flight Research Center
- Armstrong, Neil Alden (1930–2012)
- army
- Army Ballistic Missile Agency
- arm’s-length price
- Arnauld, Antoine (1612–94)
- Arndt–Eisert systhesis
- Arneb
- Arnhem, Battle of (September 1944)
- Arnold, Benedict (1741–1801)
- Arnsbergian
- aromatic compound
- aromaticity
- arousal
- Arowhanan
- ARP
- ARPA
- ARPANET
- Arpanet
- ARP cache
- ARPES
- ARPS
- ARP spoofing
- ARQ